Безусловно, самые дорогие препараты — те, которые создаются для людей с неизлечимыми или смертельными заболеваниями со сложной морфологией. Например, компания Novartis при описании стоимости своих препаратов делает акцент на их клиническом значении, влиянии на пациентов и систему здравоохранения в целом, а также на существенном улучшении качества жизни.
Лекарства от рассеянного склероза
В начале 2019 года чиновники из FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов в США — «Хайтек») разрешили распространять препарат компании Novartis — «Майзент», предназначенный для лечения различных форм рассеянного склероза, включая клинически изолированный синдром, рецидивирующий ремиттирующий рассеянный склероз (RRMS) и активный вторичный прогрессирующий рассеянный склероз (SPMS).
Рассеянный склероз ― потенциально инвалидизирующее заболевание центральной нервной системы. Это аутоиммунное заболевание, при котором поражается миелин белого вещества головного и спинного мозга. «Склероз» происходит от слова «рубец», именно они как очаги видны на МРТ при этом диагнозе.
Симптомы рассеянного склероза существенно варьируются. Некоторые люди с тяжелым течением заболевания могут потерять способность ходить и есть, в то время как другие могут испытывать длительные периоды ремиссии без каких-либо новых симптомов.
Ремиттирующий рассеянный склероз — наиболее распространенное течение заболевания. Он характеризуется четко выраженными приступами новых или нарастающих неврологических симптомов. Эти приступы, также называемые рецидивами или обострениями, сопровождаются периодами частичного или полного выздоровления (ремиссиями). Во время ремиссий, все симптомы могут исчезнуть, или некоторые симптомы могут становиться постоянными. Однако явного прогрессирования заболевания в периоды ремиссии не наблюдается. Приблизительно у 85% людей с РС первоначально диагностируется именно РРС.
Большинство людей с диагнозом РРС в срок от 3–5 лет с появления первых симптомов приходят к вторичному прогрессирующему течению болезни, при котором со временем происходит ухудшение неврологических функций и инвалидизация пациента.
«Майзент», при очень простой схеме приёма — всего один раз в день, стал первым более чем за 15 лет препаратом, специально предназначенным для людей с активным вторичным прогрессирующим склерозом. Ожидается, что годовая цена лечения им составит $88 500.
Лечебное воздействие «Майзента» заключается в секвестировании определенных белых кровяных клеток в лимфатических узлах. Именно они играют одну из ключевых ролей при воспалительной реакции, вызванной РС. Если они не будут затрагивать миелин, то не смогут вызывать чрезмерное воспаление. В третьей фазе исследования препарата, в котором приняли участие 1651 человек, доля пациентов с подтвержденным прогрессированием инвалидности была статистически значимо ниже в группе «Майзента», чем в группе с плацебо. Пациенты, получавшие препарат, имели относительное снижение частоты рецидивов на 55%.
В 2010 году FDA одобрило препарат Gilenya, ставший широко применяемым для лечения РС. Месячный курс данного препарата стоит $2500.
Novartis даже подала иск в прошлом году, пытаясь заблокировать продажи непатентованных версий Gilenya после истечения срока действия основного патента на препарат в августе. Однако пока судебная тяжба ни к чему не привела.
«Этот препарат проникает через гематоэнцефалический барьер. — объясняет доктор Тимоти Уэст, невролог из Калифорнийского университета в Сан-Франциско. — Результаты показывают, что он замедляет прогрессирование РС у людей с более поздней стадией заболевания. При рассеянном склерозе пораженные клетки попадают в мозг и разрушаются изнутри. Это лекарство действует непосредственно в мозге, не оказывая колоссального влияния на работу других систем организма».
Но, несмотря на заверения группы экспертов, первая группа пациентов столкнулась с побочными эффектами. Как оказалось, «Майзент» может вызвать сильные головные боли, повышение артериального давления, повышение уровня печеночных ферментов и уменьшение количества лейкоцитов крови.
В марте 2019 года FDA также утвердило препарат «Мавенклад» (действующее вещество — кладрибрин), предназначенный для рецидивирующего ремиттирующего рассеянного склероза (RRMS) и активного вторичного прогрессирующего рассеянного склероза (SPMS). Представители компании сообщили, что годовая стоимость двухлетнего курса лечения составит $99 500.
В США около миллиона человек живут с РС, в России — порядка 150 тыс. человек с подтвержденным диагнозом РС, а во всем мире этим недугом страдают 2,3 млн человек. Последним методом лечения РС, одобренным FDA, был окрелизумаб в 2017 году. Это анти-CD20 моноклональное антитело, поражающее зрелые B-лимфоциты, являясь иммуносупрессором. Он вышел на рынок с заявленной стоимостью $65 000 за год лечения.
Лечение от спинальной мышечной атрофии
Та же швейцарская компания Novartis представила в апреле 2019 года препарат «Зольгенсма». Это генная терапия, которая лечит наследственное состояние, называемое спинальной мышечной атрофией. СМА — редкое генетическое заболевание. Его причиной является мутация в гене SMN1 (ген выживаемости моторных нейронов — «Хайтек»). Ген кодирует синтез белка SMN, который присутствует во всем организме и отвечает за питание и функцию моторных нейронов — специализированных нервных клеток. Лечение направлено на дефектный ген, так как моторные нейроны головного и спинного мозга контролируют работу мышц всего тела. Нехватка белка SMN приводит к гибели моторных нейронов, в результате чего наступает мышечная слабость, вплоть до инвалидности и летального исхода. СМА поражает одного из 6–10 тыс. рождённых детей каждый год.
Механизм действия «Зольгенсма» прицельно воздействует на саму причину заболевания. Функциональная копия гена SMN доставляется c помощью вирусного вектора на основе адено-ассоциированного вируса к моторным нейронам. Препарат вводится один раз и вызывает экспрессию белка SMN в моторных нейронах. Это приводит к улучшению функции мышц и общего состояния ребенка.
Безопасность и эффективность препарата оценивались на основании двух исследований, одно из которых уже завершилось. Всего в них приняли участие 36 пациентов с инфантильной СМА. Возраст детей на момент начала исследований — от двух недель до восьми месяцев. Первичные доказательства эффективности основаны на данных о состоянии 21 пациента, которых лечили препаратом «Зонгельсма».
По сравнению с естественным течением болезни, пациенты, которых лечили препаратом, демонстрировали значительные улучшения моторного развития — дети смогли держать головку и самостоятельно сидеть.
Но стоимость «Зонгельсма» зашкаливает — целых $2,1 млн за одно введение. В ответ на нападки со стороны прессы и общественности Вас Нарасимхан, исполнительный директор Novartis, сказал, что этот препарат является «историческим достижением» и «знаковой единовременной генной терапией», потому вполне справедливо является таким дорогостоящим. При этом, по его словам, стоимость «Зонгельсмы» составляет всего половину цены существующих методов лечения пациентов с СМА, при учете дополнительных трат на аппараты для поддержки дыхания и вспомогательные средства. Стоимость хронической терапии пациентов, страдающих СМА, в действительности может превышать $4 млн в первые 10 лет жизни ребенка, потому, возможно, если препарат способен не вылечить, но продлить ребенку способность сохранять подвижность — такое лечение можно считать своего рода экономией.
Утраченное зрение
В 2018 году компания Spark Therapeutics представила свой препарат «Лукстурна», возвращающий зрение пациентам, потерявшим его вследствие наследственных патологий сетчатки. Цена медицинского чуда: $425 000 за один глаз, то есть для большинства больных общая сумма составит $850 000. «Лукстурна» — это геннотерапевтический препарат, который может излечить редкую разновидность слепоты — ретинальную дистрофию, появляющаяся в результате наследования мутации в нефункциональной форме гена RPE65 от обоих родителей.
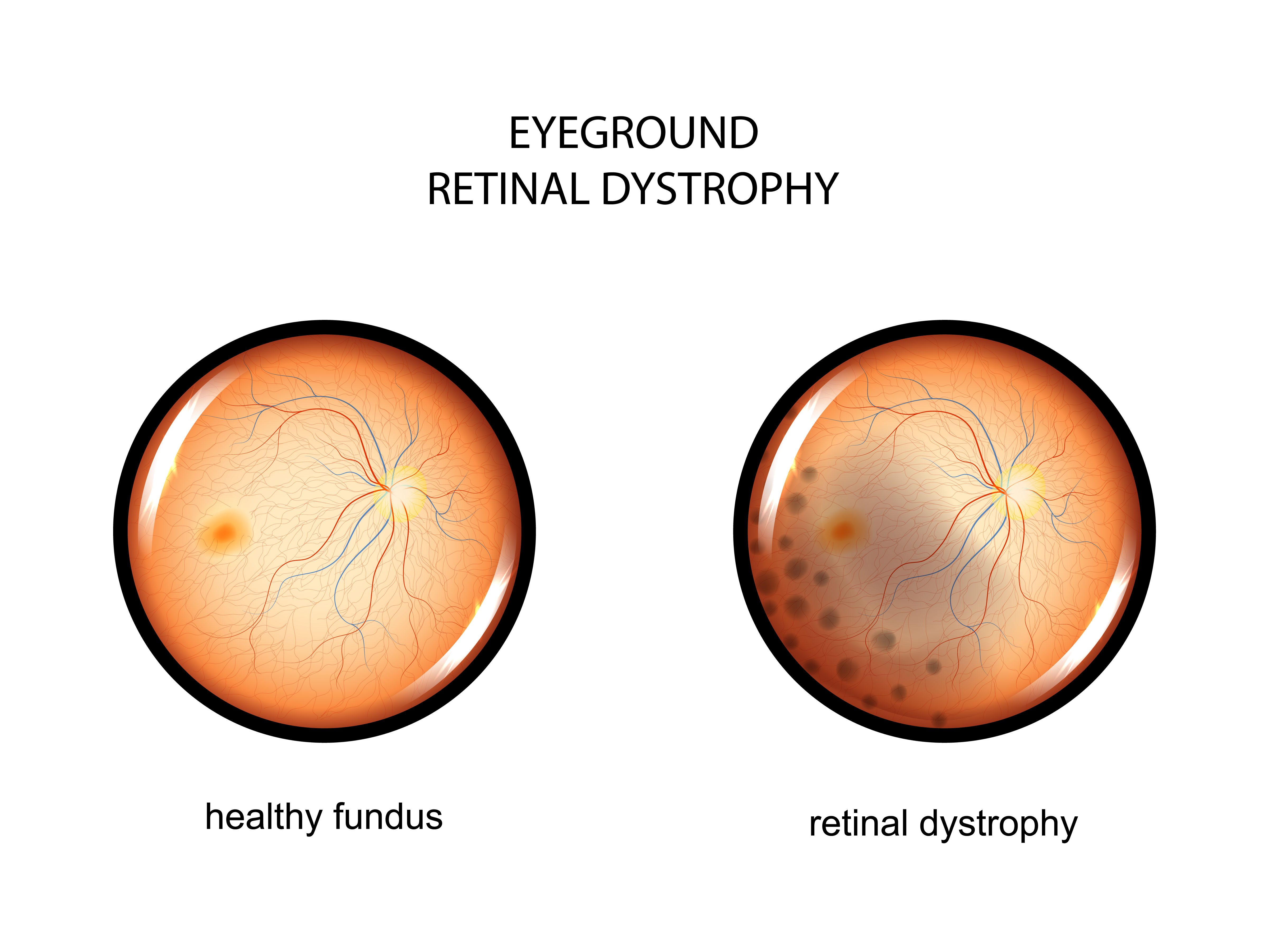
Ген RPE65 кодирует специфичный фермент клеток сетчатки глаза, участвующий в регенерации светочувствительного пигмента, который необходим для палочек и колбочек, чтобы обеспечить человеку нормальное зрение. Мутации в этом гене приводят к снижению или отсутствию уровней активности фермента, блокируя зрительный цикл и приводя к ухудшению зрения. Существует несколько типов наследственных заболеваний сетчатки, связанных с вышеназванным геном. Наиболее распространенными считают врожденный амавроз Лебера и пигментный ретинит. Из-за биаллельных мутаций гена RPE65 (генных мутаций обоих родителей) страдающие этими заболеваниями часто сталкиваются с никталопией (ночной слепотой) из-за снижения светочувствительности в детском или раннем взрослом возрасте и нистагмом (непроизвольными движениями зрачков). По мере прогрессирования заболевания происходят потеря периферического зрения, развитие туннельного зрения и, в конечном итоге, потеря центрального зрения, что означает полную слепоту.
Ретинальная дистрофия или наследственные дегенеративные заболевания сетчатки — это целый ряд болезней, которые отличаются по своей патологии, симптомам, а также последствиями. Очень трудно предсказать, насколько сильно ухудшится зрение, и как быстро будет прогрессировать заболевание, но во многих случаях люди с наследственной дистрофией сетчатки могут полностью потерять свое зрение.
Врожденный амавроз Лебера (ВАЛ) — редкое наследственное заболевание, которое приводит к нарушению деятельности сетчатки глаза и ухудшению зрения в раннем детстве, часто прямо с рождения. Из всех заболеваний, связанных с дегенерацией сетчатки, ВАЛ начинается раньше всего и может иметь наиболее тяжкие последствия. Уровень потери зрения при ВАЛ различается от случая к случаю, но остается стабильным в 75% случаев. У примерно 15% детей отмечается прогрессирующая потеря зрения, а у 10% может отмечаться некоторое незначительное, часто временное, улучшение.
Пигментный ретинит — группа наследственных заболеваний сетчатки, которые приводят к постепенному прогрессирующему снижению зрения. Потеря бокового зрения (иначе известного как «туннельное зрение») и снижение способности видеть в ночное время являются первыми заметными симптомами ПР. Далее страдающие этим заболеванием могут испытывать снижение зрения при чтении (детальное зрение), а затем — потерю цветового и центрального зрения.
Текущие методы лечения таких болезней включают генную терапию, клеточную терапию и протезирование сетчатки. Генная терапия имеет наибольший потенциал для достижения окончательного излечения путем замены мутационного гена. Терапия при помощи препарата «Лукстернa» доставляет нормальную копию гена RPE65 непосредственно в клетки сетчатки, которые затем вырабатывают нормальный белок. Он и преобразует свет в электрический сигнал в сетчатке для восстановления потери зрения пациентов. «Лукстерна» создана на основе встречающегося в природе аденоассоциированного вируса, который был модифицирован с использованием методов рекомбинации ДНК.
Аденоассоциированный вирус — малый вирус, инфицирующий клетки человека и некоторых других приматов. Не приводит к возникновению заболевания у человека и, соответственно, вызывает слабый иммунный ответ. Может инфицировать делящиеся и неделящиеся клетки и встраивать свой геном в геном хозяина. Эти особенности делают AAV особенно привлекательным кандидатом для создания вирусных векторов для генной терапии.
Иное применения средства от рака
«Актиммун» (интерферон гамма-1b) изначально был разработан компанией Horizon Pharma как препарат для лечения рака — это рекомбинантный белок, похожий на тот, что вырабатывается иммунной системой человека — интерферон-гамма. Тесты «Aктиммун» в качестве потенциального лечения онкобольных людей в сочетании с другими препаратами все еще продолжаются.
Но FDA уже одобрило его для лечения двух редких заболеваний — хронического гранулематозного заболевания и тяжелого злокачественного остеопороза.
Хроническое гранулематозное заболевание — наследственное заболевание, при котором иммунная система отсутствует или не функционирует должным образом.
Тяжелый злокачественный остеопороз — «мраморная» болезнь, которая влияет на нормальный рост здоровых костей.
Как и «оригинальный» интерферон, «Aктиммун» является сигнальным белком, который связывается с рецепторами клеточной поверхности. Это приводит к транскрипции генов, которые активируют определенные клетки в иммунной системе. Препарат подталкивает антигены, связанные со злокачественной опухолью, к воздействию на клетки иммунной системы. В целом ожидается, что «Актиммун» будет усиливать ответ иммунной системе, что приведет к гибели опухолевых клеток.
Наиболее распространенными побочными эффектами при приеме «Актиммуна» компания-производитель называет симптомы, напоминающие грипп — лихорадку, головную боль, озноб и быструю утомляемость, которые могут снижаться по мере продолжения лечения. Но риск побочных эффектов куда более незначителен по сравнению с объективной пользой препарата. Уже доказано, что он замедляет течение как мраморной болезни, так и хронического гранулематоза. Стоимость искусственного аналога интерферона-гаммы составляет около $572 тыс. в год.
Помощь пациентам с синдромом Хантера
Синдром Хантера представляет собой рецессивное наследственное заболевание, вызванное недостаточным уровнем в организме лизосомального фермента. Проще говоря, необходимый для правильного углеводного обмена фермент не вырабатывается в нужных количествах и полисахариды (естественные сахара) неправильно усваиваются. В связи с отсутствием или наличием дефектного фермента у пациентов с синдромом Хантера полисахариды накапливаются в лизосомах клеток, приводя к клеточному перенасыщению, органомегалии, разрушению тканей и нарушению функций внутренних органов. Частота заболевания в мире составляет один человек на 100 тысяч. В России зарегистрировано около 150 пациентов.
Идурсульфаза — это очищенная форма лизосомального фермента идуронат-2-сульфатазы, полученная на линии клеток человека, обеспечивающей профиль гликозилирования, аналогичный естественному ферменту, которого не хватает у пациентов с болезнью Хантера. Это вещество и стало активным в препарате американской фармацевтической компании «Шайер Хьюман Дженетик Терапис» — «Элапраза», первом и пока единственном, который может существенно облегчить жизнь страдающим от этого нарушения людям. «Элапраза» вводится внутривенно, а курс лечения подразумевает ежедневные инфузии на протяжении всей жизни.
Безопасность и эффективность препарата были оценены в клиническом исследовании контрольной группы, состоявшей из 96 пациентов с синдромом Хантера. Пациенты, получавшие еженедельное лечение продемонстрировали значительное улучшение по сравнению с теми, кто принимал плацебо. У пациентов в возрасте от 16 месяцев до пяти лет «Элапраза» не дала существенного улучшения симптомов, связанных с заболеванием, или отдаленного клинического результата. Однако лечение значительно уменьшило размер селезенки маленьких пациентов так же, как и у детей в возрасте 5 лет и старше. Стоимость «Элапразы» составляет $375 тыс. в год.
В 2011 году Куйбышевский суд Петербурга обязал Комитет по здравоохранению пожизненно обеспечивать больного синдромом Хантера Юру Веселова препаратом «Элапраза». До этого, несмотря на многочисленные жалобы матери мальчика и уполномоченного по правам ребенка в Петербурге Светланы Агапитовой, комитет отказывался предоставить льготное право на получение лекарства, ежегодный курс которого на тот момент оценивался в 30 млн рублей.
Наследственный ангионевротический отек
Наследственный ангионевротический отек (HAE) — очень редкое и потенциально смертельное заболевание, которое встречается примерно у одного из 10 тыс. человек в мире. Главный симптом HAE — отеки в различных частях тела. У пациентов часто бывают приступы мучительной боли в животе, тошнота и рвота, вызванные отеком кишечной стенки. Отек дыхательных путей особенно опасен и может привести к смерти от удушья.
У пациентов с НАЭ имеется дефект в гене, который контролирует белок крови, называемый ингибитором С1. Генетический дефект приводит к выработке либо неадекватного, либо нефункционального белка C1-Inhibitor. Нормальный C1-Inhibitor помогает регулировать сложные биохимические взаимодействия систем на основе крови, участвующих в борьбе с болезнями, воспалительной реакцией и коагуляцией. Поскольку дефектный C1-Inhibitor не выполняет надлежащим образом свою регуляторную функцию, может возникнуть биохимический дисбаланс и продуцировать нежелательные пептиды. Они побуждают капилляры высвобождать жидкость в окружающую ткань, вызывая тем самым отек.
«Цинриз» является первым ингибитором C1-эстеразы (C1-INH), одобренным FDA для предотвращения приступов внезапных отеков у детей (от шести лет и старше), подростков и взрослых. Этот препарат с 2008 года успешно используется для снижения частоты, тяжести и продолжительности отеков у подростков и взрослых пациентов, страдающих ангиодистрофией. Было показано, что у детей в возрасте от шести до 11 лет «Цинриз» уменьшает количество и степень тяжести приступов наследственных ангионевротических отеков и практически избавляет от необходимости использования неотложной терапии. Он вводится внутривенно каждые 3–4 дня для предотвращения или смягчения приступов. Эффективность и безопасность «Цинриза» были оценены в 36-недельном клиническом испытании у 12 детей с HAE в возрасте от семи до одиннадцати лет. Пациентов наблюдали в течение 12 недель для установления базовой частоты приступов, затем давали одну дозу препарата (500 или 1000 ед. каждые 3–4 дня) в течение 12 недель, а затем уменьшали или увеличивали дозу в течение последних 12 недель. Эффективность измеряли в зависимости от уменьшения количества приступов при изменении дозы лекарства по сравнению с исходным уровнем. Стоимость годового курса «Цинриз» — $1,1 млн.
В 2018 году у «Цинриза» появилась альтернатива — детище той же компании «Шайер» — препарат «Тахзиро», стоимость которого — $20 тыс. за одну дозу.
Стоимость жизненно важных препаратов зависит от множества исходных данных, но и их польза неоспорима. Обеспечить идентичную замену дефектным клеткам или способствовать выработке нужных для больного ферментов, зачастую могут только сложные разработки, в которых задействованы большое количество специалистов. В России, к большому сожалению, еще не все эти лекарства одобрены или выдаются по квоте всем нуждающимся. Но однажды мы к этому придем.